Abiomed (Nasdaq:ABMD) annonce deux approbations de la Food and Drug Administration (FDA) des États-Unis liées à la recherche clinique des pompes cardiaques Impella chez les patients atteints d’infarctus aigu du myocarde (IAM) en état de choc cardiogénique.
Ce communiqué de presse contient des éléments multimédias. Voir le communiqué complet ici : https://www.businesswire.com/news/home/20220916005096/fr/
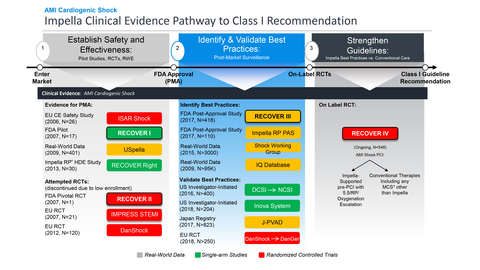
RECOVER IV is an on-label, two-arm RCT that is designed to provide the clinical evidence needed to achieve a Class I guideline recommendation for Impella use in AMI cardiogenic shock. (Graphic: Business Wire)
La FDA a approuvé l’essai randomisé contrôlé (ERC) RECOVER IV sur étiquette pour les patients atteints de choc cardiogénique IAM. RECOVER IV est un essai à deux volets qui évaluera si l’intervention coronarienne percutanée (ICP), avec prise en charge Impella initiée avant l’ICP, est supérieure à l’ICP sans prise en charge Impella.
« Cet essai historique sera l’aboutissement de plus de 20 ans de recherche dans le traitement interventionnel de l’IAM et mettra en œuvre toutes les avancées cliniques que nous avons réalisées pour améliorer la survie et la récupération cardiaque des patients atteints d’IAM en état de choc cardiogénique, comme l’ont démontré de multiples études prospectives », déclare William O’Neill, MD, directeur médical du Center for Structural Heart Disease chez Henry Ford Health et co-chercheur principal national de RECOVER IV.
Le critère d’évaluation principal de RECOVER IV est la mortalité toutes causes confondues à 30 jours. Les critères d’évaluation secondaires comprennent les événements indésirables cardiovasculaires et cérébrovasculaires majeurs (MACCE) à 30 jours, les jours de vie hors de l’hôpital à six mois, la récupération de la fonction ventriculaire gauche (LV), le besoin d’un dispositif d’assistance ventriculaire durable (VAD) ou d’une transplantation cardiaque et la qualité de vie liée à la santé, mesurée par les réponses au questionnaire sur la cardiomyopathie de Kansas City (KCCQ) à un an. L’objectif d’Abiomed dans la conduite de cet essai est d’obtenir une recommandation mondiale de classe I sur les chocs cardiogéniques pour Impella et les protocoles de bonnes pratiques associés, y compris l’implantation d’Impella pré-ICP (voir figure 1).
« Je suis optimiste quant au fait que RECOVER IV démontrera plus encore les avantages de la prise en charge hémodynamique et des protocoles de bonnes pratiques. Ces avantages incluent la décharge ventriculaire à l’aide de l’Impella pré-ICP, la réduction du stress de la paroi LV, la réduction de la congestion pulmonaire, l’augmentation du débit sanguin coronaire collatéral et l’amélioration de la protection cardio afin que davantage de patients atteints de choc cardiogénique IAM puissent survivre et réussir une récupération cardiaque native. L’équipe et le domaine cardiaques ont évolué et comprennent à quel point la récupération du myocarde est importante pour l’IAM et le choc cardiogénique de l’IAM afin de réduire l’épidémie croissante d’insuffisance cardiaque », explique Navin K. Kapur, MD, directeur exécutif du Cardiovascular Center for Research and Innovation (CVCRI) au Tufts Medical Center et co-chercheur principal national pour RECOVER IV.
La FDA approuve et clôture l’étude post-approbation RECOVER III
De plus, la FDA a approuvé et clôturé l’étude post-approbation (PAS) prospective sur les chocs cardiogéniques d’Impella, RECOVER III. Cette étude a rassemblé des preuves concrètes sur des patients atteints de choc cardiogénique IAM traités avec Impella entre 2017 et 2019, en recueillant des données détaillées, notamment sur les stades du choc cardiogénique, le débit cardiaque et le moment de l’implantation. RECOVER III répond à l’exigence PAS d’Abiomed et l’approbation et la clôture de RECOVER III par la FDA valident en outre Impella comme thérapie sûre et efficace pour le choc cardiogénique IAM.
Impella reste le seul dispositif d’assistance circulatoire mécanique (MCS) à avoir reçu le plus haut niveau d’approbation préalable à la mise sur le marché (PMA) et d’approbation réglementaire PAS de la FDA pour le choc cardiogénique IAM. Sur la base de RECOVER III, l’étiquette d’Impella pour le choc cardiogénique IAM sera mise à jour pour refléter les données jusqu’à un an après la procédure.
Antécédents cliniques de choc cardiogénique IAM
Le choc cardiogénique IAM a l’un des taux de mortalité les plus élevés dans le domaine de la médecine. Le taux de survie est resté d’environ 50 % pour les patients en état de choc cardiogénique aux stades D et E du SCAI sans prise en charge Impella et les bonnes pratiques associées. La survie seule en cas de choc cardiogénique n’est plus la référence. Plusieurs études sur les bonnes pratiques Impella démontrent une survie supérieure à 70 % avec une récupération cardiaque native supérieure à 90 % (voir figure 2). Les bonnes pratiques d’Impella, telles que l’implantation pré-ICP (voir figure 3), ont été développées par des médecins experts reconnus dans le domaine de l’assistance circulatoire et publiées au cours de la dernière décennie dans de multiples études cliniques aux États-Unis, en Allemagne, en Italie et au Japon. La récupération cardiaque après un choc cardiogénique IAM améliore la qualité de vie des patients et fait d’Impella l’une des thérapies les plus rentables dans la population CMS Medicare et dans l’assurance privée. Rien qu’aux États-Unis, plus de 200 000 patients sont admis à l’hôpital chaque année en état de choc cardiogénique.
Antécédents réglementaires de la FDA concernant le choc cardiogénique IAM
Impella est la pompe cardiaque la plus étudiée de l’histoire de la FDA (voir figures 4 et 5) et possède une PMA exclusive de la FDA en tant que thérapie sûre et efficace pour le choc cardiogénique, l’ICP à haut risque et l’insuffisance cardiaque droite. Depuis 2004, plus de 1 200 études évaluées par des pairs, y compris des analyses de preuves réelles, des études cliniques prospectives et des ERC ont été publiées sur les avantages cliniques d’Impella.
Impella a été utilisé pour traiter plus de 235 000 patients dans le monde et est inclus dans 13 directives de sociétés cliniques. En 2021, la Société européenne de cardiologie a fait passer Impella à une recommandation de classe IIa pour le traitement du choc cardiogénique. Le ballonnet intra-aortique (IAB) est actuellement de classe III (nocif) pour une utilisation de routine en cas de choc cardiogénique en Europe et au Japon sur la base de l’ERC IABP-SHOCK II, qui a démontré que l’IAB n’apportait aucun avantage pour la survie ou l’augmentation hémodynamique par rapport à la thérapie inotrope. En 2020, l’IAB est devenu de classe III (nocif) dans les recommandations des lignes directrices américaines pour le choc cardiogénique post-cardiotomie.
Tous les MCS et dispositifs d’assistance ventriculaire (VAD) depuis 1992 ont été approuvés avec des études à un seul volet par rapport aux taux de survie historiques jugés sur des critères de performance objectifs (OPC) en raison des défis éthiques et logistiques liés à la randomisation des patients gravement malades nécessitant une augmentation hémodynamique immédiate. En 2008 et 2009, Abiomed a tenté l’ERC RECOVER II de la FDA, qui comparait Impella à l’IAB dans le choc cardiogénique IAM. RECOVER II n’a inscrit qu’un seul patient en 15 mois dans plus de 30 sites avant d’être interrompu pour des problèmes de consentement logistique et éthique et un manque de volontaires.
La FDA a accordé l’autorisation 510(k) à Impella en 2008 et après plusieurs études FDA et prospectives initiées par des médecins, la FDA a approuvé l’ICP à haut risque en 2015, le choc cardiogénique IAM en 2016 (voir figure 6) et d’autres formes d’insuffisance cardiaque avec choc cardiogénique en 2018. Avec l’achèvement de RECOVER III et l’approbation de l’ERC RECOVER IV, Abiomed poursuit une étude conforme pour renforcer les directives mondiales et améliorer les résultats pour les patients.
Abiomed a parrainé et financé plusieurs études sur les chocs cardiogéniques IAM depuis 2006 (voir figure 7), y compris les seules études de la FDA dans ce domaine. La difficulté de la randomisation des patients atteints d’un choc cardiogénique en cours d’exécution a été démontrée dans de nombreuses études, notamment « IMPRESS in STEMI » (n = 18), « IMPRESS in Cardiac Arrest » (n = 48), Seyfarth et al. (n = 26) et l’ERC RECOVER II de la FDA parrainé par Abiomed (n = 1). Toutes ces études n’ont pas pu être randomisées et ont été interrompues prématurément pour ne pas avoir inscrit leurs chiffres désignés en raison de problèmes de consentement logistiques et éthiques.
Modifications réglementaires récentes de la FDA pour l’exception au consentement éclairé (EFIC)
La randomisation des patients dans l’AMICS a été difficile, car ils ont besoin de soins d’urgence et sont trop malades pour fournir un consentement éclairé traditionnel pour s’inscrire à un essai. En 1996, la FDA a créé l’exception de la voie du consentement éclairé (EFIC) pour la recherche clinique d’urgence. Cette voie permet aux chercheurs d’éduquer largement une communauté sur un essai, puis d’inscrire des patients sans le consentement des patients, de leur famille ou de leurs représentants légalement autorisés.
En 2022, après avoir collaboré avec la FDA sur la conception de l’ERC RECOVER IV, la FDA a approuvé le protocole d’étude ERC RECOVER IV, qui inclut l’utilisation de l’EFIC. Ce processus de sensibilisation communautaire est rare et n’est utilisé que lorsque les patients étudiés souffrent d’une condition médicale potentiellement mortelle entraînant une déficience grave de la fonction mentale. Il s’agit d’une étape importante pour le domaine et ses principaux médecins. Les prochaines étapes comprennent les approbations du comité d’examen institutionnel (IRB) de l’hôpital local et un engagement des médecins à randomiser.
« Cet essai randomisé crucial est historique, car il est le premier à employer le consentement de la communauté EFIC pour recruter des patients en choc cardiogénique. Je salue la FDA pour sa collaboration qui aide à résoudre les problèmes de consentement dans les ERC de choc cardiogénique et j’appelle la communauté des médecins à recruter et à randomiser les patients dans le cadre de RECOVER IV », déclare Gregg W. Stone, MD, professeur de médecine et directeur des affaires académiques pour le Mount Sinai Heart Health System à New York et chaire d’étude pour RECOVER IV.
Pour plus d’informations sur les bonnes pratiques actuelles dans le traitement des patients atteints de choc cardiogénique IAM, veuillez cliquer ici.
Pour plus d’informations sur l’ERC RECOVER IV, veuillez cliquer ici.
À PROPOS DES POMPES CARDIAQUES IMPELLA
Impella 2.5® and Impella CP® with SmartAssist® are U.S. FDA approved to treat certain advanced heart failure patients undergoing elective and urgent percutaneous coronary interventions (PCI), such as stenting or balloon angioplasty, to reopen blocked coronary arteries.
Impella 2.5® et Impella CP® avec SmartAssist® sont approuvés par la FDA américaine pour traiter certains patients atteints d’insuffisance cardiaque avancée subissant des interventions coronariennes percutanées (ICP) électives et urgentes, telles que la pose d’un stent ou l’angioplastie par ballonnet, pour rouvrir les artères coronaires bloquées.
À PROPOS D’ABIOMED
Basé à Danvers, Massachusetts, États-Unis, Abiomed est un fournisseur de technologie médicale leader du marché qui fournit des solutions d’assistance circulatoire et d’oxygénation. Nos produits sont conçus pour permettre au cœur de se reposer en améliorant le flux sanguin et/ou en fournissant une oxygénation suffisante aux personnes souffrant d’insuffisance respiratoire. Pour plus d’informations, rendez-vous sur : http://www.abiomed.com/.
DÉCLARATIONS PROSPECTIVES
Toute déclaration prospective est soumise à des risques et incertitudes tels que ceux décrits dans les rapports périodiques d’Abiomed déposés auprès de la Securities and Exchange Commission. Les résultats réels peuvent différer sensiblement des résultats prévus.
Le texte du communiqué issu d’une traduction ne doit d’aucune manière être considéré comme officiel. La seule version du communiqué qui fasse foi est celle du communiqué dans sa langue d’origine. La traduction devra toujours être confrontée au texte source, qui fera jurisprudence.
Consultez la version source sur businesswire.com : https://www.businesswire.com/news/home/20220916005096/fr/
